


Two Northumbria University Fashion students have had their looks selected to be part of the…

In this article originally written for The Conversation*, from Northumbria University, Elliott…

An academic in architecture at Northumbria University has led the design of a ground-breaking…
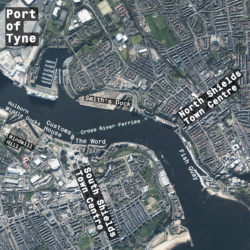
Researchers at Northumbria University have undertaken a project exploring sustainable planning…

In this article originally written for The Conversation*, Matthew T. Johnson, Professor of…

In this article originally written for The Conversation*, Intigam Mamedov, Postdoctoral research…

Two Northumbria University cyber security students have been shortlisted at the globally recognised…

Roisin Currie, Chief Executive Officer (CEO) of Greggs PLC, has been awarded an honorary degree…

Newcastle Business School
-
Lecture Theatre 022
-
Back to top


